编辑推荐:
这篇综述探讨了草本植物积雪草(CA)在对抗铝诱导的神经毒性中的潜力。研究揭示了CA通过多种机制发挥神经保护作用,包括调节氧化应激(MDA、GSH、SOD)、减轻DNA损伤、抑制细胞凋亡(Bax/Bcl2、Caspase-3)与炎症(Il-1β)相关基因表达,并通过调控GSK-3β信号通路减少β-淀粉样蛋白和Tau蛋白沉积。体内大鼠模型证实了CA能有效改善脑组织病理变化,为治疗神经退行性疾病(如阿尔茨海默病)提供了新的植物药理学见解。
全球有超过5000万人受到神经退行性疾病的困扰,阿尔茨海默病(AD)是其中最常见的类型。这类疾病以特定神经元群的逐渐丧失为特征,导致记忆、认知、感觉和运动功能的衰退。寻找有效的治疗方法至关重要。传统草药积雪草(CA,又称Gotu kola)因其富含三萜类和酚类化合物,并具有促进记忆的传统用途而受到关注。本综述重点介绍了一项旨在阐明CA提取物作为神经保护剂,对抗氯化铝(AlCl3)诱导的大鼠神经退行性变潜在分子机制的研究。
研究设计与核心发现
该研究将48只雄性成年Wistar大鼠分为八组,包括对照组、单独AlCl3处理组、三个不同剂量(200、400、600 mg/kg)的CA单独处理组,以及三个CA与AlCl3联合处理组。所有处理均通过口服持续5周。研究评估了脑组织中的氧化应激生物标志物(MDA、GSH、SOD)和DNA损伤程度,并检测了凋亡和炎症相关基因(Bax/Bcl2、Caspase3、Il-1β、Gsk3β)的mRNA表达,以及β-淀粉样蛋白和Tau蛋白的免疫组织化学染色。
积雪草的多重神经保护机制
- 1.
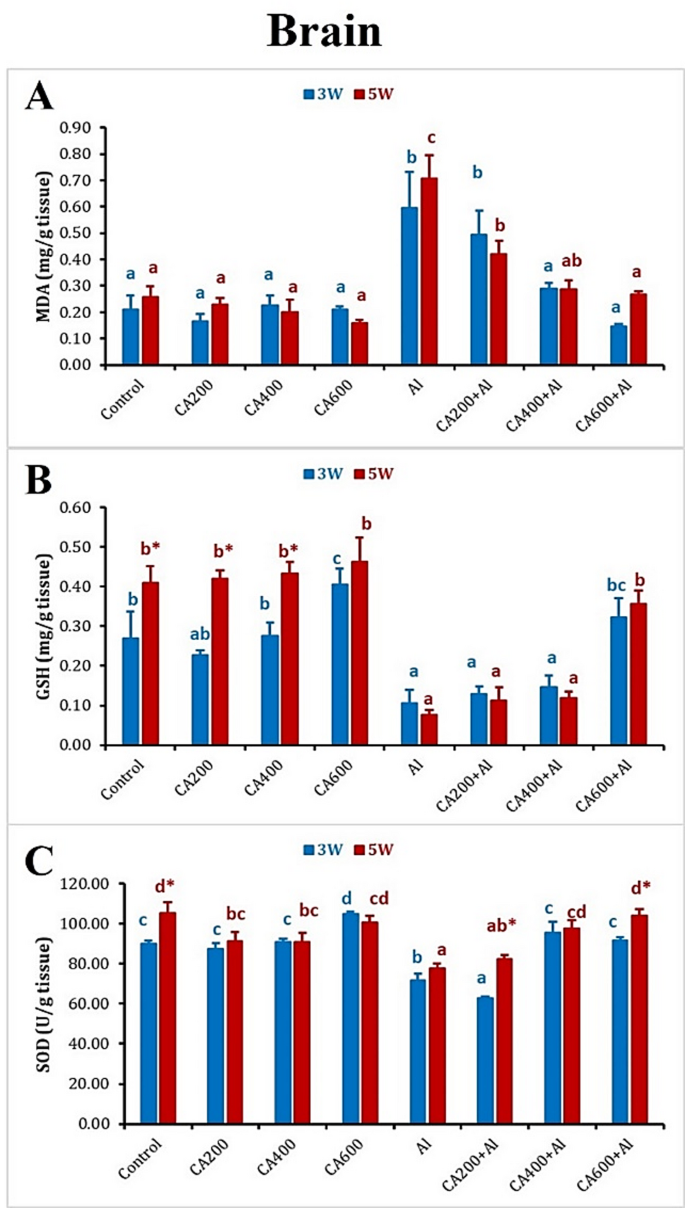
改善氧化应激与DNA损伤:AlCl
3处理导致脑组织中丙二醛(MDA)水平显著升高,而抗氧化酶谷胱甘肽(GSH)和超氧化物歧化酶(SOD)的活性降低,DNA损伤标志物(如彗星实验中的尾长、尾矩)也显著增加。而CA提取物,特别是高剂量(600 mg/kg)与AlCl
3联用,能够剂量依赖性地逆转这些变化,提升GSH和SOD水平,降低MDA和DNA损伤参数,恢复了氧化还原平衡。
- 2.
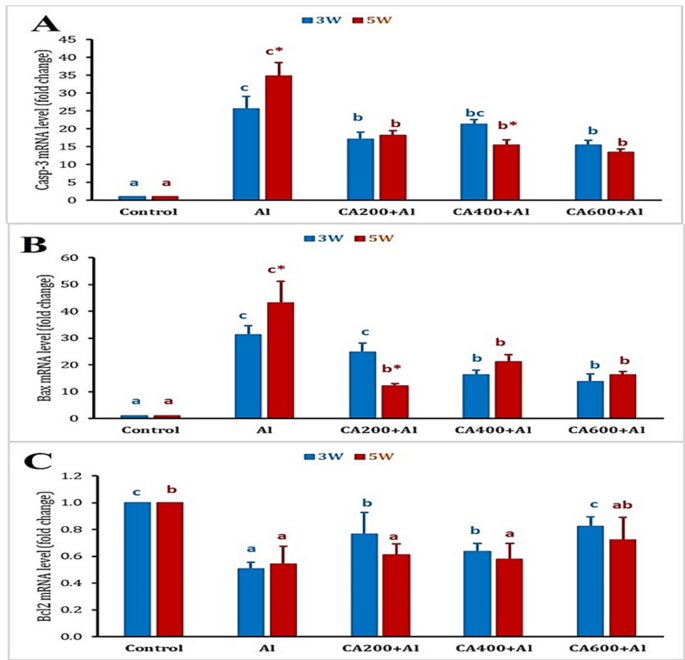
抑制炎症与细胞凋亡:AlCl
3诱导了炎症基因
Il-1β和促凋亡基因
Bax、
Caspase-3的上调,同时下调了抗凋亡基因
Bcl2。CA处理则能有效下调
Il-1β、
Bax和
Caspase-3的表达,并上调
Bcl2的表达,表明CA具有显著的抗炎和抗凋亡效应。
- 3.
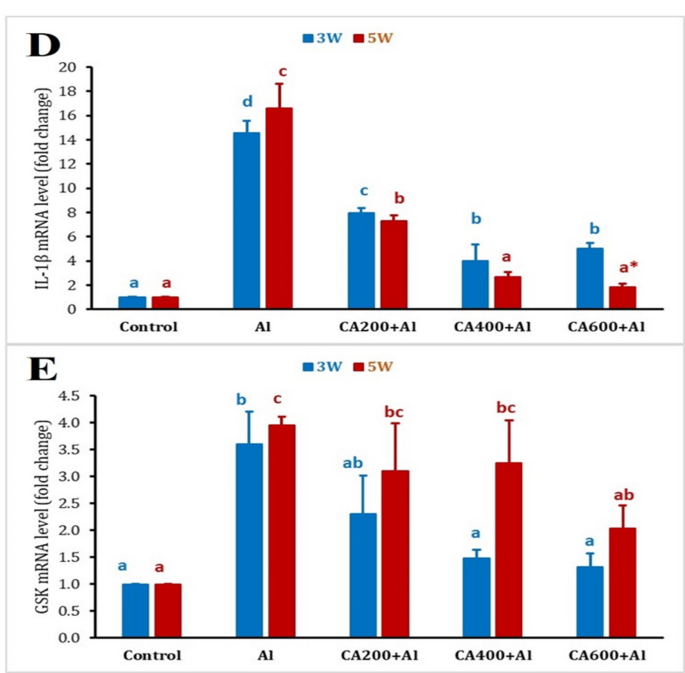
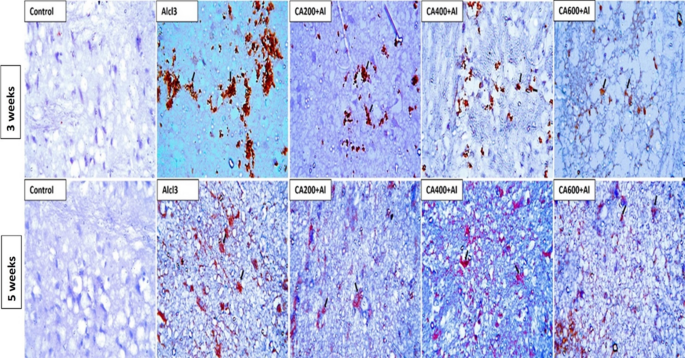
调控GSK-3β信号通路,减少病理蛋白沉积:研究的一个关键发现是CA能够下调糖原合酶激酶-3β(
Gsk-3β)的表达。GSK-3β是AD病理中的关键激酶,参与Tau蛋白的过度磷酸化和β-淀粉样蛋白(Aβ)的产生。正如预期,AlCl
3处理增加了脑内Aβ斑块和Tau蛋白的积累。而CA的干预,尤其是高剂量,能够剂量依赖性地显著减少这两种病理蛋白的沉积,这与
Gsk-3β的下调密切相关,提示CA可能通过调节GSK-3β通路来发挥其神经保护作用。
- 4.
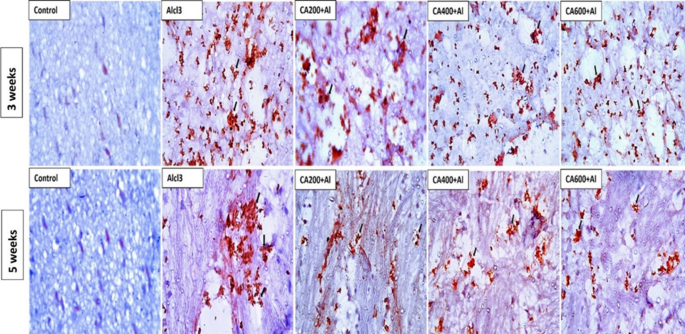
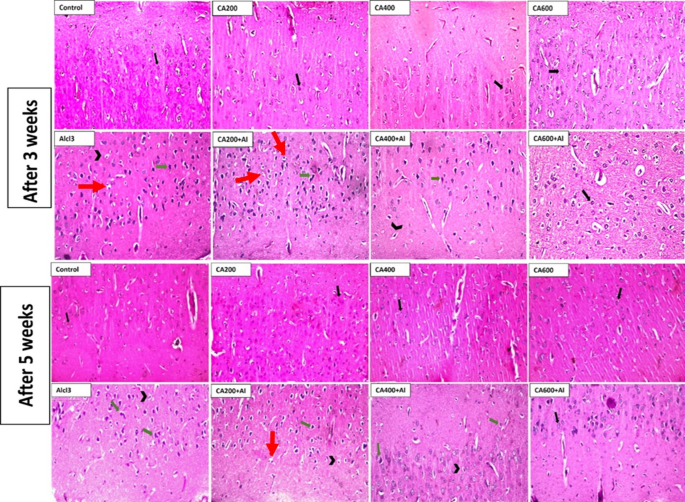
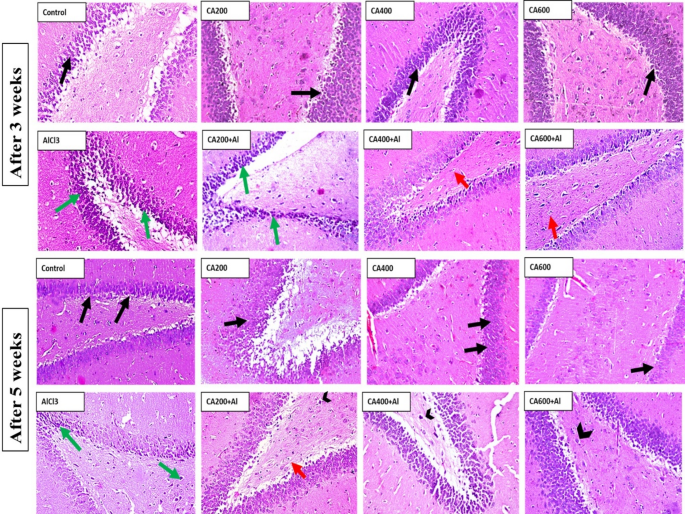
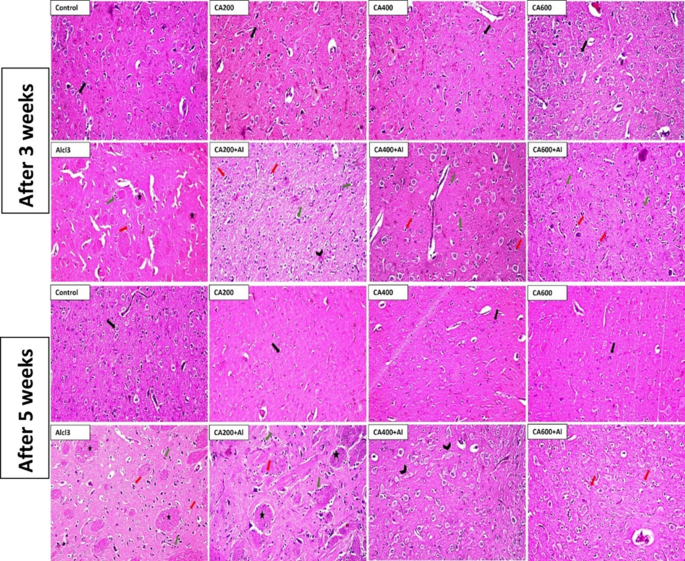
改善脑组织病理学结构:组织病理学检查进一步证实了CA的保护作用。在AlCl
3单独处理组中,大鼠大脑皮层、海马和纹状体均出现了神经元核固缩、胶质增生、变性和局灶性嗜酸性斑块形成等严重病变。而在CA与AlCl
3联合处理组中,这些病变得到明显改善,且改善程度呈剂量依赖性,CA600联合处理组的脑组织形态最接近正常对照组。
结论与展望
综上所述,积雪草(CA)展现出了对抗铝诱导神经毒性的强大、多靶点神经保护潜力。其机制网络涵盖:增强内源性抗氧化防御、修复DNA损伤、遏制神经炎症与细胞凋亡级联反应,并通过调控核心的GSK-3β信号通路,有效减少AD特征性病理蛋白Aβ和Tau的积累,从而在组织形态学上显著减轻脑损伤。这些发现为将CA及其活性成分(如积雪草苷)开发为预防或治疗神经退行性疾病(尤其是与环境和代谢毒素相关的认知障碍)的潜在植物药或膳食补充剂,提供了扎实的临床前药理学依据。未来的研究需要进一步分离和鉴定其具体活性成分,在更特异的疾病模型中进行验证,并系统评估其长期使用的安全性。











